






KAIST(총장 신성철)가 슈퍼 컴퓨팅을 이용해 암세포 유형별로 최적의 약물표적을 발굴하는 기술을 개발했다. 일부가 아닌 모든 환자에게 쓰일 수 있는 암 관련 신약 개발을 촉진시킬 것으로 기대된다.
KAIST는 조광현 바이오 및 뇌공학과 교수팀이 암세포의 유전자 변이가 반영된 분자네트워크 다이나믹스(동역학)를 분석, 유형별 최적의 약물표적을 발굴하는 기술을 개발했다.
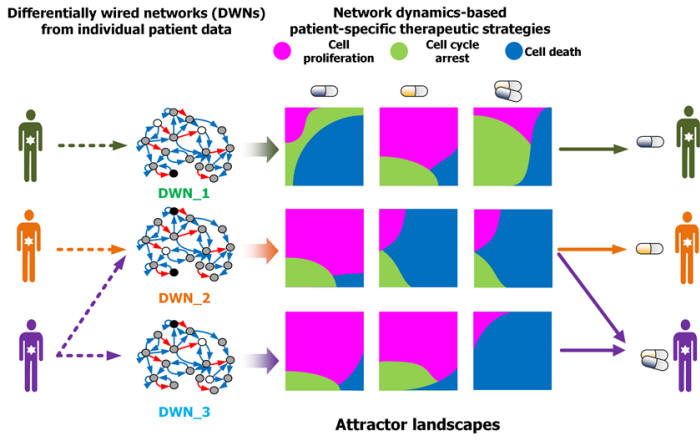
인간의 암세포는 유전자 돌연변이, 유전체단위의 반복적 변이를 비롯해 다양한 변이를 거친다. 이런 변이는 같은 암종에서도 암세포에 따라 많은 차이를 보인다. 약물 반응도 가지각색이다.
특정 약물에 대응하는 유전자변이를 파악하는 것이 신약 개발에 중요하지만 기존의 연구 방법으로는 한계가 있었다. 그동안 학계가 암세포의 유전자 변이에 관한 분자네트워크 동역학 특성에 대해 큰 관심을 두지 않았기 때문이다. 소수의 암 관련 유전자를 표적으로 둬, 대다수의 치료법이 일부 환자에게만 유용했다.
연구팀은 슈퍼컴퓨팅을 이용한 대규모 시뮬레이션과 세포실험을 융합해 약물반응 예측 기술을 개발했다. 막대한 계산능력으로 암세포 분자네트워크의 동역학 변화를 분석했다. 시범적으로 대다수 암 발생에 관여하는 것으로 알려진 암 억제 유전자 'p53'을 분석하는 것에도 성공했다.

연구팀은 '암 세포주 백과사전(CCLE)'에 공개된 대규모 암세포 유전체 데이터를 분자네트워크에 반영, 서로 다른 유전변이 특성을 가진 분자네트워크를 생성했다. 이후 약물반응을 모사하는 '섭동분석'을 통해 약물반응에 따른 암세포의 변화를 정량화, 군집화 했다.
연구팀은 이 기술로 다양한 암세포 약물반응의 원인을 파악할 수 있다고 설명했다. 특정 유전자나 단백질 뿐만아니라 이들의 상호 조절 작용도 함께 고려해 분석할 수 있다. 최적의 약물표적을 발굴하고 기존 약물의 새로운 적용대상을 창출할 수 있다.
조광현 교수는 “암세포 유형별 분자네트워크의 약물반응을 시뮬레이션으로 분석했다”면서 “약물 반응 근본 원리를 파악하고 새로운 최적 약물 타켓을 발굴할 수 있게 됐다”고 말했다.
대전=김영준기자 kyj85@etnews.com

